Différences entre les versions de « VBTutorial1 »
| Ligne 9 : | Ligne 9 : | ||
The Gamess and XMVB input files for the H<math>{}_2</math> molecule are provided in the ''Exercise'' folder on the tutorial machines. These are VBSCF calculations with the 6-31G(d,p) basis set, with the fragment specification in terms of symmetry-adapted orbitals (''frgtyp=sao''). | The Gamess and XMVB input files for the H<math>{}_2</math> molecule are provided in the ''Exercise'' folder on the tutorial machines. These are VBSCF calculations with the 6-31G(d,p) basis set, with the fragment specification in terms of symmetry-adapted orbitals (''frgtyp=sao''). | ||
| − | Just inspect these inputs, run the gamess-xmvb program (using : ''vbrun h2''), and analyze the outputs. | + | Just inspect these inputs, run the gamess-xmvb program (using : ''vbrun h2-scf''), and analyze the outputs. |
Then these input files could serve you as templates for the next exercises. | Then these input files could serve you as templates for the next exercises. | ||
Version du 12 juillet 2012 à 08:43
Basics of VB theory and XMVB program
Exercise 1 : Starting up with the H<math>{}_2</math> molecule
The Gamess and XMVB input files for the H<math>{}_2</math> molecule are provided in the Exercise folder on the tutorial machines. These are VBSCF calculations with the 6-31G(d,p) basis set, with the fragment specification in terms of symmetry-adapted orbitals (frgtyp=sao).
Just inspect these inputs, run the gamess-xmvb program (using : vbrun h2-scf), and analyze the outputs.
Then these input files could serve you as templates for the next exercises.
Exercise 2 : HF molecule weights
- Compute a VBSCF three structure wave function for the HF molecule, using the frgtyp=sao specification, automatic guess (guess=auto), and boys keyword in the $tctrl section. Which structure(s) should be kept in further BOVB calculations ?
- Using VBSCF orbitals as guess orbitals :
- Compute a L-BOVB wave function on a selected subset of structures ;
- Compute a VBCISD wave function, freezing the 1s core orbital of fluorine in the VBCI calculation (NCOR=1 option), and printing only structures which have a coefficient superior to 0.01 (ctol=0.01 option) ;
- Compare structure weights at the VBSCF, L-BOVB and VBCI levels
| Hints |
|---|
|
>> general guidelines for BOVB calculations
Exercise 3 : F<math>{}_2</math> molecule and bond energy
- Compute a L-VBSCF wave function for the F<math>{}_2</math> molecule (all inactive orbitals localized on the fluorine atoms), using:
- the frgtyp=sao specification, without using the f basis functions in the definition of the fragment orbitals for simplicity ;
- the boys keyword in the $ctrl section ;
- automatic guess (guess=auto option) ;
- Recompute the same L-VBSCF wave-function, this time specifying converged RHF MOs as guess orbitals, through the guess=mo option in the $ctrl section together with an extra $gus section in the input (see hints below, XMVB Manual, and/or Peifeng Su's lecture slides) ;
- BOVB level :
- First, compute a π-D-VBSCF wave function using previous VBSCF orbitals as guess orbitals. To do that, you should allow the π inactive orbitals of fluorine to delocalize onto the two atoms, while keeping all <math>\sigma</math> (active and inactive) orbitals localized (see also : >> see "high symmetry case" in the "general guidelines for BOVB calculations")
- Compute then a π-D-BOVB solution for the F<math>{}_2</math> molecule, starting from previous orbitals as guess.
- VBCI : compute a VBCI(D,S) wave function (vbcids keyword in the $ctrl section), freezing the core orbitals of fluorine in the calculation.
- Deduce F<math>{}_2</math> bond energies at both the π-D-BOVB and VBCI(D,S) levels.
| Hints and remarks |
|---|
|
To prepare a $gus section for reading RHF MOs as a guess (guess=mo option) :
Note that using automatic guess works fine in a simple case like this one, using guess=mo simply accelerate convergence. However, for larger molecule, specifying a good orbital guess through guess=mo and an extra $gus section will often be useful. For VBCI(D,S) calculation on difluorine : don't forget to add NCOR=2 and ctol=0.01 options in the $Ctrl section. To compute the bond energies :
Note that a more accurate BOVB bond energy could be obtained by pushing to higher SD-BOVB level, and with VBCISD by using a larger basis set. |
Exercise 4 : The lone pairs of H2O
(for further reading, see S. Shaik and P.C. Hiberty, "The Chemist's Guide to VB theory", Wiley, Hoboken, New Jersey, 2008, pp. 107-109)
This exercise aims at comparing two descriptions of the lone pairs of H<math>{}_2</math>O : (i) the MO description in terms of non-equivalent canonical MOs and (ii) the « rabbit-ear » VB description in terms of two equivalent hybrid orbitals.

- Focusing on the lone pairs only, write the four-electron single-determinants <math>\Psi_{\textrm{MO}} </math> and <math>\Psi_{\textrm{VB}} </math> .
- Expand <math>\Psi_{\textrm{VB}} </math> into elementary determinants containing only <math>n</math> and <math>p</math> orbitals, eliminate determinants having two identical spinorbitals, and show the equivalence between <math>\Psi_{\textrm{VB}}</math> and <math>\Psi_{\textrm{MO}}</math>.
- We now remove one electron from H<math>{}_2</math>O. Write the two possible VB structures <math>\Phi_1</math> and <math>\Phi_2</math> in the VB framework. By convention, one may write the doubly occupied lone pair first, then the singly occupied one.
- The two ionized states are the symmetry-adapted combinations
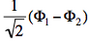 and
and 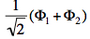 . From the sign of the hamiltonian matrix element <math>\langle \Phi_1 \vert \hat{H} \vert \Phi_2 \rangle</math>, give the energy ordering of the two ionized states.
. From the sign of the hamiltonian matrix element <math>\langle \Phi_1 \vert \hat{H} \vert \Phi_2 \rangle</math>, give the energy ordering of the two ionized states. - By expanding the two ionized states into elementary determinants (dropping the normalization constants), show that they are equivalent, respectively, to the MO configurations <math>\vert nn\bar{p}\vert</math> and <math>\vert pp\bar{n}\vert</math>.
| Appendix |
|---|
|
Hamiltonian matrix element between determinants differing by one spin-orbital : Don’t forget to put the orbitals of the two determinants in maximal correspondence before applying the rule. |
| Answer |
|---|
|
1.
<math> \Psi_{\textrm{MO}}=\vert n\bar{n}p\bar{p}\vert </math> ; <math> \Psi_{\textrm{VB}}=\vert \left( n-\lambda p\right)\left( \bar{n}-\lambda\bar{p}\right)\left( n+\lambda p\right)\left(\bar{n}+\lambda\bar{p}\right)\vert </math>
2.
<math> \begin{matrix} \Psi_{\textrm{VB}} &=&\vert \left( n\bar{n} -\lambda p\bar{n} -\lambda n\bar{p} +\lambda^2 p\bar{p} \right) \left( n\bar{n} +\lambda p\bar{n} +\lambda n\bar{p} +\lambda^2 p\bar{p} \right) \vert \end{matrix} </math> <math> \begin{matrix} \Psi_{\textrm{VB}} &=&\vert n\bar{n}n\bar{n} \vert + \lambda\vert n\bar{n}p\bar{n} \vert + \lambda\vert n\bar{n}n\bar{p} \vert + \lambda^2 \vert n\bar{n}p\bar{p} \vert - \lambda \vert p\bar{n}n\bar{n} \vert -\lambda^2 \vert p\bar{n}p\bar{n} \vert -\lambda^2 \vert p\bar{n}n\bar{p} \vert -\lambda^3 \vert p\bar{n}p\bar{p} \vert \end{matrix} </math> <math> \begin{matrix} - \lambda \vert n\bar{p}n\bar{n} \vert -\lambda^2 \vert n\bar{p}p\bar{n} \vert -\lambda^2 \vert n\bar{p}n\bar{p} \vert -\lambda^3 \vert n\bar{p}p\bar{p} \vert + \lambda^2 \vert p\bar{p}n\bar{n} \vert +\lambda^3 \vert p\bar{p}p\bar{n} \vert +\lambda^3 \vert p\bar{p}n\bar{p} \vert +\lambda^4 \vert p\bar{p}p\bar{p} \vert \end{matrix} </math> After eliminating all determinants having two orbitals with the same spin, there remains : <math> \begin{matrix} \Psi_{\textrm{VB}} &=& \lambda^2 \vert n\bar{n}p\bar{p} \vert -\lambda^2 \vert p\bar{n}n\bar{p} \vert -\lambda^2 \vert n\bar{p}p\bar{n} \vert + \lambda^2 \vert p\bar{p}n\bar{n} \vert \end{matrix} </math> After permuting the columns and changing signs accordingly, there remains : <math> \Psi_{\textrm{VB}}=4\lambda^2\vert n\bar{n}p\bar{p} \vert=\Psi_{\textrm{MO}} </math> (if one includes normalization factors).
3.
Permuting the first and third orbitals in <math>\Phi_2</math> and changing the sign accordingly, we get <math>-\Phi_2</math> that has maximum orbital correspondence with <math>\Phi_1</math> : <math>-\Phi_2</math> =<math>\vert \left( n-\lambda p \right) \left( \bar{n}+\lambda\bar{p} \right) \left( n+\lambda p \right) \vert </math>.
4.
5.
<math>\Phi_1=+2\lambda^2\vert p\bar{p}n \vert +2\lambda\vert n\bar{n}p \vert</math> In the same way, one shows that <math>\Phi_2=-2\lambda^2\vert p\bar{p}n \vert +2\lambda\vert n\bar{n}p \vert</math>. It follows that : <math>\left(\Phi_1-\Phi_2\right)\propto \vert n\bar{n}p \vert </math> (lowest ionized state in MO theory) <math>\left(\Phi_1+\Phi_2\right)\propto \vert p\bar{p}n \vert </math> (higher ionized state in MO theory). It is concluded that 1) VB theory yields two ionization potentials for H<math>{}_2</math>O, in agreement with experiment, and 2) that these ionization potentials are exactly the same as the ones found in elementary MO theory.
|