VBTutorial3
How to modify this page :
- first : log in (top right of this page) ;
- click on [edit] (far right) to edit a section of the page ;
- write your text directly in the wiki page, and click on the "Save page" button (bottom left) to save your modifications
Pictures : how to insert a picture in your text
See also this page for an introduction to the basics of the wiki syntax
Remarks
Exercises
Main Exercise
Exercise 1 : Computation of state correlation Diagrams for a 3 centers / 4 electrons system
- Computer exercise :
In this exercise the <math>\textrm{S}_{\textrm{N}}2</math> reaction Cl<math>{}^{-}</math> + CH3Cl -> ClCH3 + Cl<math>{}^{-}</math> will be computed in both vacuum and solution, at <math>\pi</math>-D-BOVB level. The basis set is 6-31+G*, for there is an anoin in the system. To make the computation simplified, only reactant and transition state will be computed in the exercise. It is recommended to the advanced users to draw the whole VBSCD.
- Compute VBSCD diagrams for Cl(-) + CH3Cl -> ClCH3 + Cl(-), at π-D-BOVB levels, first in gas phase then using VB(PCM)... Which basis set should we use : 6-31+G*. As this is an anion we should add a set of diffuse functions, but then there may be trouble with BOVB... Check first that everything is fine at BOVB level (no instability)...
- Reactant and product geometry will be CH3Cl and Cl(-) computed separately (CH3Cl has already been computed in tutorial 1, we just have to change the basis set, which is easy with XMVB 2.0 !) ;
- TS transition state : we take the linear TS, with orientation of the x or y axis along a C-H bond this will make a <math>\sigma</math>, <math>\pi_x</math>, <math>\pi_y</math> separation, so we could apply π-D-BOVB level, localizing all <math>\sigma</math> pairs (including inactive <math>\sigma</math> lone pairs and cores of Carbon and Chlorine atoms, and delocalizing the and <math>\pi_x</math> and <math>\pi_y</math> inactive orbitals from the beginning ((see also "high symmetry cases" here)|.
>> general guidelines for BOVB calculations
Exercise 2 : computation of H—H + H. -> H. + H—H radical exchange VBSCD diagram
1/ Paper exercise :

a/ Considering the following radical exchange process: <math> X^{\bullet} + A-Y \rightarrow X-A + ^{\bullet}Y </math>
Write the HL wave functions for R and R* and derive the value of G using semiempirical VB theory.
b/ Considering the following reaction:
<math>
X^{\bullet} + H-X \rightarrow X-H +^{\bullet}X
</math>
Use semiempirical VB theory to derive the following expression for the avoided cross term B:
<math>
B=0.5 BDE
</math> where BDE is the Bond Dissociation Energy.
c/ Use semiempirical VB theory to show why the reaction: <math> X^{\bullet} + H-X \rightarrow X-H + ^{\bullet}X</math> has a barrier for <math>X= CH_{3}</math>, <math>SiH_{3}</math>, <math>GeH_{3}</math>, <math>SnH_{3}</math>, <math>PbH_{3}</math>, <math>H</math>, while the <math>Li_{3}</math> species in the process <math> Li^{\bullet} + Li-Li \rightarrow Li-Li + ^{\bullet}Li</math> is a stable intermediate.
First construct a VBSCD with the usual parameters <math>\Delta E_{ST}, f, G, B</math>.
Where <math>\Delta E_{ST}</math> is the singlet-triplet transition energy of the X-H bond at the geometry of the transition state. For convenience, define the energy of a Lewis bond, for example, H-X (or X-X), relative to the nonbonded quasiclassical reference determinant, as follows:
<math>E_{S}(H-X)=-\lambda_{S}</math> and <math>D(H-X)=\lambda_{S}</math> where <math>\lambda_{S}</math> is used as a shorthand notation for <math>2\beta S/(1+S^2)</math>.
Similarly, denote the energy of the triplet pair <math>H \uparrow \uparrow X</math> (or of <math>X \uparrow \uparrow X</math>) by: <math>E_{T}(H \uparrow \uparrow X)=\lambda_{T}</math>, where <math>\lambda_{T}</math> is the corresponding <math>-2\beta S/(1-S^2)</math> terms for the triplet repulsion. While <math>\lambda_{S}</math> and <math>\lambda_{T}</math> are defined at the equilibrium distance of the H-X (or X-X) bond, analogous parameters <math>\lambda'_{S}</math> and <math>\lambda'_{T}</math> are defined for the stretched H-X (or X-X) bond corresponding to the geometry of the transition state. Based on these notations derive the following relations and quantities:
- Express <math>\Delta E_{ST}</math> and <math>G</math> as functions of <math>\lambda_{S}</math> and <math>\lambda_{T}</math>.
- Express the avoided crossing interaction <math>B</math> as a function of <math>\lambda'_{S}</math> and <math>\lambda'_{T}</math>.
- Express the energy of the crossing point (relative to the reference quasiclassical determinant) as a function of <math>\lambda'_{S}</math> and <math>\lambda'_{T}</math>.
- To enable yourself to derive a simple expression for the barrier, assume that <math>\lambda'_{S}=\lambda'_{T}</math>. Then express <math>\Delta E_{C}</math>, the height of the crossing point relative to the reactants, and derive an expression for <math>f</math>, as a function of <math>\alpha</math>, defined as follows: <math>\alpha=\lambda_{S}/\lambda_{T}</math>.
- Derive an expression for the barrier <math>\Delta E^{\neq}</math>, as a function of <math>\Delta E_{ST}</math> and <math>\alpha</math>. Knowing that <math>\lambda_{T}</math> is generally larger than <math>\lambda_{S}</math> for strong binders, such as X-H (<math>X= CH_{3}</math>, <math>SiH_{3}</math>, <math>GeH_{3}</math>, <math>SnH_{3}</math>, <math>PbH_{3}</math>, <math>H</math>) while the opposite is true for weak binders, such as alkali atoms, show that the reaction <math>X^{\bullet} + H-X \rightarrow X-H + ^{\bullet}X</math> has a barrier while the <math>Li_{3}</math> species is a stable intermediate in the process <math> Li^{\bullet} + Li-Li \rightarrow Li-Li + ^{\bullet}Li</math>.
2/ Computer exercise :
idea : Compute VBSCD diagrams for H—H + H. -> H. + H—H at VBSCF then VBCI level.
Exercise 3 (paper exercise) : Conical intersection in H3• radical
(for further reading, see S. Shaik and P.C. Hiberty, "The Chemist's Guide to VB theory", Wiley, Hoboken, New Jersey, 2008, pp. 157-161, exercises 6.11-6.14 pp. 174-176, and answers to the exercises pp. 188-192.
Consider three hydrogen atoms Ha, Hb, Hc, with respective atomic orbitals a, b and c, and the two VB structures ![]() ] and
] and ![]() ] .
] .
The Ha-Hb and Hb-Hc distances are equal. ![]()
- By using the thumb rules recalled below, where squared overlap terms are neglected, derive the expression of the energies of R and P, and of the reduced Hamiltonian matrix element between R and P for the 3-orbital/3-electrons reacting system [Ha--Hb--Hc]•.
- From the sign of this latter integral when θ > 60°, derive the expressions of the ground state Ψ≠ and of the first excited state Ψ* of the H3• system. One may drop the normalization constants for simplicity. What bonding scheme does the excited state represent ?
- Show that the reduced Hamiltonian matrix element is largest in the collinear transition state geometry, and drops to zero in the equilateral triangular structure.
- Show that R and P VB structures are degenerate in the equilateral triangular structure, and that Ψ≠ and Ψ* are also degenerate in this geometry.
- We now extend the above conclusions to the allyl radical. What are the bonding schemes corresponding to the ground state and first excited state ? What geometrical distortion would make these two states degenerate ? What would be the end product of a photochemical excitation of allyl radical to its first excited state ?
Appendix : Thumb rules for the calculations of effective Hamiltonian matrix elements between determinants.
- Energy of a determinant D :
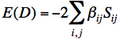 ] (if orbitals i and j have parallel spins)
] (if orbitals i and j have parallel spins) - Matrix element between determinants differing by spin inversion of two spin-orbitals :
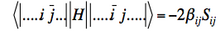 ]
]