VBTutorial3
How to modify this page :
- first : log in (top right of this page) ;
- click on [edit] (far right) to edit a section of the page ;
- write your text directly in the wiki page, and click on the "Save page" button (bottom left) to save your modifications
Pictures : how to insert a picture in your text
See also this page for an introduction to the basics of the wiki syntax
Remarks
Exercises
>> general guidelines for BOVB calculations
Exercise 1 : computation of H—H + H. -> H. + H—H radical exchange VBSCD diagram
1/ Paper exercise :

a/ Considering the following radical exchange process: <math> X^{\bullet} + A-Y \rightarrow X-A + ^{\bullet}Y </math>
Write the HL wave functions for R and R* and derive the value of G using semiempirical VB theory.
b/ Considering the following reaction:
<math>
X^{\bullet} + H-X \rightarrow X-H +^{\bullet}X
</math>
Use semiempirical VB theory to derive the following expression for the avoided cross term B:
<math>
B=0.5 BDE
</math> where BDE is the Bond Dissociation Energy.
c/ Use semiempirical VB theory to show why the reaction: <math> X^{\bullet} + H-X \rightarrow X-H + ^{\bullet}X</math> has a barrier for <math>X= CH_{3}</math>, <math>SiH_{3}</math>, <math>GeH_{3}</math>, <math>SnH_{3}</math>, <math>PbH_{3}</math>, <math>H</math>, while the <math>Li_{3}</math> species in the process <math> Li^{\bullet} + Li-Li \rightarrow Li-Li + ^{\bullet}Li</math> is a stable intermediate.
First construct a VBSCD with the usual parameters <math>\Delta E_{ST}, f, G, B</math>.
Where <math>\Delta E_{ST}</math> is the singlet-triplet transition energy of the X-H bond at the geometry of the transition state. For convenience, define the energy of a Lewis bond, for example, H-X (or X-X), relative to the nonbonded quasiclassical reference determinant, as follows:
<math>E_{S}(H-X)=-\lambda_{S}</math> and <math>D(H-X)=\lambda_{S}</math> where <math>\lambda_{S}</math> is used as a shorthand notation for <math>2\beta S/(1+S^2)</math>.
Similarly, denote the energy of the triplet pair <math>H \uparrow \uparrow X</math> (or of <math>X \uparrow \uparrow X</math>) by: <math>E_{T}(H \uparrow \uparrow X)=\lambda_{T}</math>, where <math>\lambda_{T}</math> is the corresponding <math>-2\beta S/(1-S^2)</math> terms for the triplet repulsion. While <math>\lambda_{S}</math> and <math>\lambda_{T}</math> are defined at the equilibrium distance of the H-X (or X-X) bond, analogous parameters <math>\lambda'_{S}</math> and <math>\lambda'_{T}</math> are defined for the stretched H-X (or X-X) bond corresponding to the geometry of the transition state. Based on these notations derive the following relations and quantities:
- Express <math>\Delta E_{ST}</math> and <math>G</math> as functions of <math>\lambda_{S}</math> and <math>\lambda_{T}</math>.
- Express the avoided crossing interaction <math>B</math> as a function of <math>\lambda'_{S}</math> and <math>\lambda'_{T}</math>.
- Express the energy of the crossing point (relative to the reference quasiclassical determinant) as a function of <math>\lambda'_{S}</math> and <math>\lambda'_{T}</math>.
- To enable yourself to derive a simple expression for the barrier, assume that <math>\lambda'_{S}=\lambda'_{T}</math>. Then express <math>\Delta E_{C}</math>, the height of the crossing point relative to the reactants, and derive an expression for <math>f</math>, as a function of <math>\alpha</math>, defined as follows: <math>\alpha=\lambda_{S}/\lambda_{T}</math>.
- Derive an expression for the barrier <math>\Delta E^{\neq}</math>, as a function of <math>\Delta E_{ST}</math> and <math>\alpha</math>. Knowing that <math>\lambda_{T}</math> is generally larger than <math>\lambda_{S}</math> for strong binders, such as X-H (<math>X= CH_{3}</math>, <math>SiH_{3}</math>, <math>GeH_{3}</math>, <math>SnH_{3}</math>, <math>PbH_{3}</math>, <math>H</math>) while the opposite is true for weak binders, such as alkali atoms, show that the reaction <math>X^{\bullet} + H-X \rightarrow X-H + ^{\bullet}X</math> has a barrier while the <math>Li_{3}</math> species is a stable intermediate in the process <math> Li^{\bullet} + Li-Li \rightarrow Li-Li + ^{\bullet}Li</math>.
2/ Computer exercise :
idea : Compute VBSCD diagrams for H—H + H. -> H. + H—H at VBSCF then VBCI level.
Exercise 2 : Computation of state correlation Diagrams for a 3 centers / 4 electrons system
- No paper exercise.
- Computer exercise :
ideas :
- Compute VBSCD diagrams for Cl(-) + CH3Cl -> ClCH3 + Cl(-), at π-D-BOVB levels, first in gas phase then using VB(PCM)... Which basis set should we use : 6-31+G*. As this is an anion we should add a set of diffuse functions, but then there may be trouble with BOVB... Check first that everything is fine at BOVB level (no instability)...
- Reactant and product geometry will be CH3Cl and Cl(-) computed separately (CH3Cl has already been computed in tutorial 1, we just have to change the basis set, which is easy with XMVB 2.0 !) ;
- TS transition state : we take the linear TS, with orientation of the x or y axis along a C-H bond this will make a <math>\sigma</math>, <math>\pi_x</math>, <math>\pi_y</math> separation, so we could apply π-D-BOVB level, localizing all <math>\sigma</math> pairs (including inactive <math>\sigma</math> lone pairs and cores of Carbon and Chlorine atoms, and delocalizing the and <math>\pi_x</math> and <math>\pi_y</math> inactive orbitals from the beginning ((see also "high symmetry cases" here)|.
Exercise 3 (paper exercise) : Conical intersection in H3• radical
(for further reading, see S. Shaik and P.C. Hiberty, "The Chemist's Guide to VB theory", Wiley, Hoboken, New Jersey, 2008, pp. 157-161, exercises 6.11-6.14 pp. 174-176, and answers to the exercises pp. 188-192.
Consider three hydrogen atoms Ha, Hb, Hc, with respective atomic orbitals a, b and c, and the two VB structures ![]() ] and
] and ![]() ] .
] .
The Ha-Hb and Hb-Hc distances are equal. ![]()
- By using the thumb rules recalled below, where squared overlap terms are neglected, derive the expression of the energies of R and P, and of the reduced Hamiltonian matrix element between R and P for the 3-orbital/3-electrons reacting system [Ha--Hb--Hc]•.
- From the sign of this latter integral when θ > 60°, derive the expressions of the ground state Ψ≠ and of the first excited state Ψ* of the H3• system. One may drop the normalization constants for simplicity. What bonding scheme does the excited state represent ?
- Show that the reduced Hamiltonian matrix element is largest in the collinear transition state geometry, and drops to zero in the equilateral triangular structure.
- Show that R and P VB structures are degenerate in the equilateral triangular structure, and that Ψ≠ and Ψ* are also degenerate in this geometry.
- We now extend the above conclusions to the allyl radical. What are the bonding schemes corresponding to the ground state and first excited state ? What geometrical distortion would make these two states degenerate ? What would be the end product of a photochemical excitation of allyl radical to its first excited state ?
Appendix : Thumb rules for the calculations of effective Hamiltonian matrix elements between determinants.
- Energy of a determinant D :
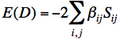 ] (if orbitals i and j have parallel spins)
] (if orbitals i and j have parallel spins) - Matrix element between determinants differing by spin inversion of two spin-orbitals :
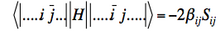 ]
]